1月28日,中國科學院上海營養與健康研究所章海兵研究組在國際學術期刊Advanced Science在線發表了題為 “Cleavage-Resistant CYLD Protects Against Autoimmune Hepatitis” 的研究論文。該研究首次揭示了去泛素化酶CYLD在自身免疫性肝炎中的關鍵調控作用,發現CYLD蛋白剪切是驅動肝臟炎癥反應的重要分子開關,并系統闡明了“CYLD–MEK1/2”信號軸在免疫介導性肝損傷中的關鍵作用機制,為自身免疫性肝炎的精準干預提供了新的理論基礎和潛在靶點。
自身免疫性肝炎是一類由免疫系統異常激活引起的肝臟炎癥性疾病,且疾病持續活動可進展為肝纖維化、肝硬化甚至肝細胞癌。當前臨床治療主要依賴免疫抑制策略,但部分患者療效有限且易復發,其分子致病機制尚未完全闡明,嚴重限制了靶向治療藥物的開發。因此,深入解析發病機制對開發新型治療策略、突破臨床困境至關重要。
研究人員在刀豆蛋白A(ConA)誘導的小鼠自身免疫性肝炎(EAH)模型中發現,疾病進程伴隨CYLD蛋白水平顯著下降及特異性剪切片段產生。該過程由Caspase-8介導,并發生在CYLD的Asp215位點。通過構建抵抗剪切的突變型CYLD(CyldD215A/D215A)小鼠,研究人員發現阻斷CYLD剪切可顯著緩解肝組織壞死和炎癥反應,增強小鼠對免疫性肝損傷的耐受能力。利用骨髓移植實驗與細胞特異性點突變小鼠進一步明確,僅在巨噬細胞中引入CYLD-D215A突變即可產生顯著保護效應,表明巨噬細胞內CYLD的穩定性是抑制自身免疫性肝炎的關鍵。
在分子機制層面,研究發現炎癥因子TNFα可誘導巨噬細胞中CYLD發生剪切,從而增強巨噬細胞對警報素S100A9的敏感性,促進招募中性粒細胞的趨化因子(CXCL1/2/3)過量表達,進而驅動中性粒細胞浸潤并加劇肝損傷。深入探索發現,CYLD通過特異性去除MEK1/2蛋白的K63多聚泛素鏈,削弱MEK1/2與上游RAF激酶的相互作用,從而抑制MEK1/2的磷酸化激活及其下游炎性基因表達。同時,研究鑒定出E3泛素連接酶TRIM25與CYLD協同調控MEK1/2的泛素化修飾。應用MEK1/2抑制劑可顯著減輕EAH小鼠的肝臟炎癥和損傷,進一步驗證了該信號軸在疾病進展中的核心作用。
綜上,該研究首次系統揭示了CYLD蛋白剪切在自身免疫性肝炎中的致病作用機制,提出了“CYLD剪切—MEK1/2激活—炎癥趨化因子過量產生”的全新調控軸線,不僅深化了對免疫介導性肝臟炎癥發生機制的認識,也為靶向CYLD穩定性及MEK信號通路的創新治療策略提供了重要理論依據。
中國科學院上海營養與健康研究所博士研究生劉涵,北京大學生命科學學院博士研究生蘇晨為該論文共同第一作者。中國科學院上海營養與健康研究所章海兵研究員,李明副研究員為該論文的共同通訊作者。該工作得到了中國科學院上海營養與健康研究所李于研究員、李虹研究員等的大力支持。該工作得到了科技部國家重點研發計劃項目、國家自然科學基金項目、上海市市級科技重大專項項目等的資助,以及中國科學院上海營養與健康研究所所級公共技術中心分析測試技術平臺、實驗動物技術平臺的支持。
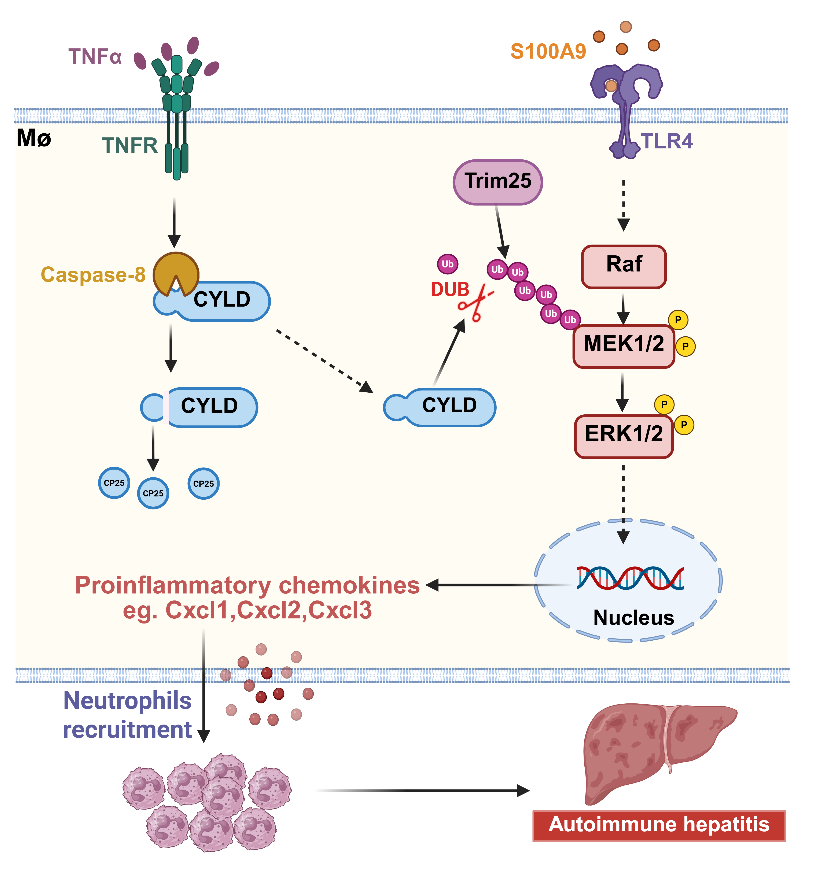
圖:CYLD-MEK1/2軸調控自身免疫性肝炎的機制示意圖
推送單元:章海兵研究組、科技規劃與任務處
 官方微信
官方微信