2023年3月1日,中國科學院上海營養與健康研究所丁秋蓉研究組在Molecular Cell雜志在線發表了題為“Histone phosphorylation integrates the hepatic glucagon-PKA-CREB gluconeogenesis program in response to fasting”的研究論文。該研究發現了組蛋白H3S28磷酸化(H3S28ph)響應饑餓情況下胰高血糖素信號,直接調控肝臟糖異生基因轉錄激活的生物學機制:在饑餓狀態下,胰高血糖素激活PKA信號通路,PKA繼而被CREB招募到基因組糖異生基因調控區域,磷酸化H3S28。磷酸化的H3S28ph被14-3-3 識別,招募更多的RNA聚合酶II(Pol II),激活糖異生基因轉錄。并發現進食狀態下,PP2A作為H3S28ph的磷酸酶抑制其磷酸化,繼而抑制糖異生基因的表達。該研究發現了組蛋白磷酸化在肝臟糖穩態調控中的生物學功能,并發現了CREB作為橋梁蛋白介導PKA磷酸化H3S28繼而調控糖異生基因轉錄的調控模式。
高血糖是2型糖尿病(T2D)的臨床特征,也是包括脂肪肝、肥胖等多種代謝性疾病的關鍵風險因素。肝臟組織在維持機體糖代謝穩態中起重要作用,而肝葡萄糖生成(HGP, hepatic glucose production)的失調會明顯加速T2D的發生和發展。肝臟HGP主要包括肝糖原的分解和糖異生。在饑餓狀態下,HGP主要來自于糖異生,而在T2D患者中,由于胰島素抵抗的發生,糖異生速率增加,進一步導致了空腹血糖的增高。靶向肝臟糖異生是治療T2D的一種潛在策略。
糖異生(Gluconeogenesis)是指非糖前體(丙酮酸、乳酸、甘油、生糖氨基酸等)轉變為糖或糖原的過程。在正常生理狀態下,饑餓時血糖迅速下降,肝臟內會發生快速的糖異生基因的表達,促進糖異生,以維持正常血糖穩態。糖異生基因包括編碼糖異生限速酶磷酸烯醇式丙酮酸羧激酶1的基因PCK1、編碼葡萄糖-6-磷酸脫氫酶的基因G6PC等,其轉錄調控主要通過CREB和Foxo1兩個關鍵轉錄因子介導的信號軸進行。然而,基因轉錄的快速啟動不僅依賴于轉錄因子的激活,組蛋白修飾改變導致的大范圍內染色質開放或閉合狀態的重塑決定了基因轉錄激活的敏感性和有效性。在饑餓狀態下糖異生基因轉錄的快速啟動過程中,染色質層面發生了什么相應的變化,至今仍不甚清晰。
丁秋蓉研究團隊首先廣泛篩選饑餓刺激的肝臟內組蛋白翻譯后修飾變化,發現H3S28ph顯著上升。結合RNA-seq和H3S28ph的ChIP-seq、CUT&Tag結果,發現饑餓會誘導H3S28ph信號的明顯增加,尤其是在糖異生相關基因的轉錄調控區域。進一步通過肝臟特異過表達野生型H3.3(H3.3-wt)和突變型H3.3(模擬磷酸化突變H3.3-S28D和非磷酸化突變H3.3-S28A),證明H3.3-S28D可以顯著促進肝臟糖異生基因表達和糖異生。
研究人員進一步探究了H3S28ph的調控機制,發現胰高血糖素可以體內、體外促進肝細胞H3S28ph的發生,并通過一系列生化實驗和體內功能實驗證明,細胞核內的PKA是H3S28ph的激酶。更有意思的是,研究人員進一步發現PKA特異結合到糖異生基因調控區域依賴于CREB的招募,PP2A是H3S28ph的磷酸酶并抑制肝臟糖異生基因的表達。
針對H3S28ph如何激活糖異生表達的機制研究,通過蛋白質譜分析發現在饑餓情況下,肝臟細胞核內的14-3-3 表達上升。進一步通過體外和CUT&Tag等實驗證明,14-3-3 是H3S28ph的表觀閱讀器,14-3-3 可以識別H3S28ph,并招募更多的Pol II到糖異生基因啟動子區域, 促進基因轉錄;而敲除肝臟14-3-3 可以明顯抑制糖異生。
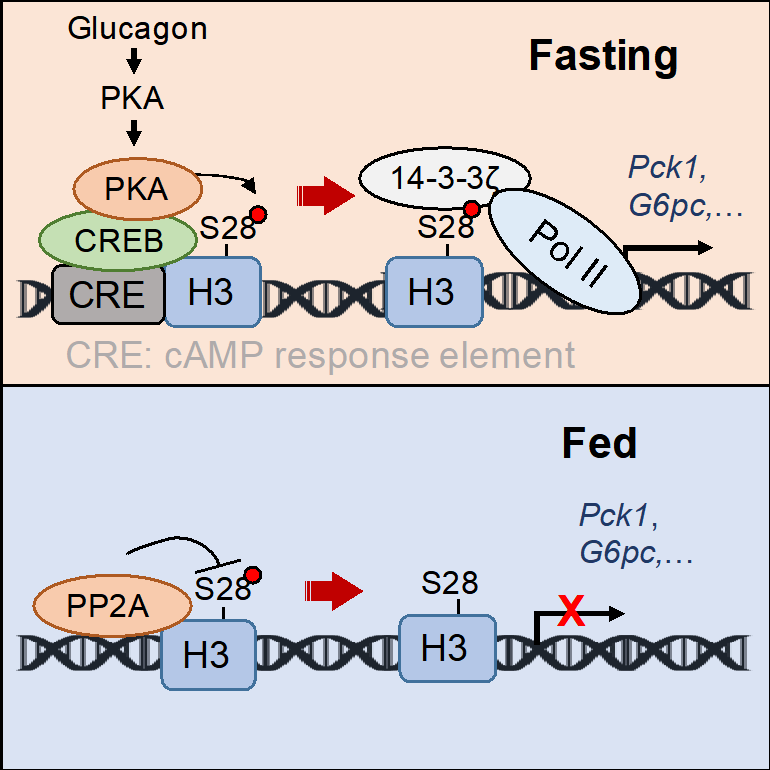
圖示. 在饑餓情況,血液內胰高血糖素上升并與肝臟上受體結合,促進肝臟細胞內cAMP上升。PKA被激活并釋放出催化亞基,與基因組上的CREB結合后促進H3S28ph。14-3-3 作為表觀閱讀器識別H3S28ph并招募RNA聚合酶II激活糖異生基因轉錄。相反地,PP2A是H3S28ph磷酸酶并抑制糖異生基因表達。
綜上所述,該研究揭示了組蛋白磷酸化修飾快速響應饑餓刺激的胰高血糖素-PKA信號通路,進而促進肝臟糖異生基因轉錄的工作機制,豐富了對機體在染色質層面如何響應激素變化,調控代謝穩態的生物學認識。
該項研究得到了復旦大學基礎醫學院劉悒教授,中科院上海營養與健康研究所陳雁、秦駿、周犇研究員,上海交通大學附屬第六人民醫院胡承教授的大力支持。中國科學院上海營養與健康研究所的副研究員趙永旭、助理研究員李爽為本文的第一作者,丁秋蓉研究員為本文通訊作者,趙永旭為共同通訊作者。該研究得到國家自然科學基金委、上海市科委、中國科學院青年創新促進會的支持,同時得到中國科學院上海營養與健康研究所公共技術中心和動物平臺的支持。
 官方微信
官方微信