2017年4月25日,國際學術期刊Cell Reports在線發表了中國科學院上海生命科學研究院(人口健康領域)章海兵研究組的最新研究成果 “RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in Fadd-deficient mice”。該研究揭示了細胞程序性壞死關鍵調控蛋白RIPK3在胚胎發育及成體炎癥調控過程中的不同作用機制。
細胞程序性死亡是多細胞生物的基本生命活動。細胞死亡調控異常與包括腫瘤、自身免疫性疾病、神經退行性疾病以及代謝性疾病在內的多種疾病相關聯。細胞壞死一直被認為是不受基因調控的細胞死亡形式。但近年隨著細胞程序性壞死被發現,關于細胞程序性壞死的分子機制及其在生理病理條件下的作用越來越受到關注。RIPK3是介導細胞程序性壞死信號通路的關鍵調控蛋白,RIPK3的缺失能完全阻斷細胞程序性壞死的發生。隨著研究的深入,不斷有研究發現RIPK3還參與炎癥小體激活以及多種炎癥信號的調控,但RIPK3在不同條件下如何決定及調控不同的信號機制還不清楚。
在章海兵研究員的指導下,博士研究生趙群等通過構建RIPK3激酶結構域突變的小鼠,發現該突變小鼠能正常的生存,體內體外實驗均證實該激酶結構域突變導致激酶活性降低并特異性的抵抗細胞程序性壞死的發生。之前的研究發現細胞凋亡基因FADD的缺失導致的胚胎致死是細胞程序性壞死依賴的,即RIPK3或MLKL的敲除能夠挽救FADD敲除小鼠的胚胎發育致死。因此我們利用該激酶結構域突變小鼠與FADD敲除小鼠交配,意外發現RIPK3激酶結構域突變與FADD雙敲除的小鼠在出生后一天死亡,并伴有腸道出血癥狀。進一步對其機制探究,發現腸道組織中TNF-a、IL-1b和CCL2等炎性因子和趨化因子的表達分泌較野生型小鼠顯著升高,體外誘導分化的巨噬細胞在LPS刺激條件下,IL-1β的表達量明顯升高,揭示FADD缺失條件下的自發性腸道炎癥,其調控不依賴于RIPK3激酶功能。此研究在遺傳水平證實,相較于RIPK3敲除能完全恢復FADD缺失導致的發育及炎癥表型,RIPK3激酶結構域突變挽救FADD敲除小鼠的胚胎發育,但不能阻斷FADD成體缺失誘導的腸道炎癥,揭示了RIPK3在胚胎發育及成體炎癥調控過程中的不同作用機制,為進一步開發針對RIPK3特異性功能的藥物提供靶點及理論依據。
該研究得到應浩研究員的支持和幫助,獲得國家自然科學基金委等項目資助。同時也得到了上海生科院(人口健康領域)公共技術平臺以及動物平臺的支持。(科技處)
?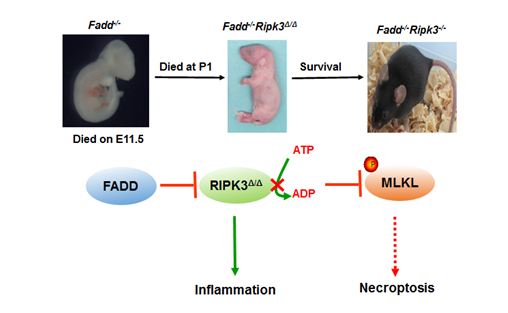
圖: RIPK3在胚胎發育及炎癥調控中的作用機制RIPK3在胚胎發育及炎癥調控中的作用機制
 官方微信
官方微信